研究紹介
背景
教室主宰者の堀は、1998年に当教室の前身である発生細胞化学教室(微生物薬品化学教室)の名取俊二先生(現名誉教授)のもとで博士(薬学)の学位を取得しました。
その後、免疫学に分野を変え、ポルトガル・リスボン郊外にあるInstituto Gulbenkian de Ciência(グルベンキアン科学研究所)のAntonio Coutinho先生、Jocelyne Demengeot先生のもとに留学し、Treg研究を始めました。
留学中は、Tregが感染症における致死的な炎症応答を阻止する一方で微生物排除を抑制してしまうこと(Hori et al., Eur J Immunol, 2002a)、Tregが自己免疫疾患の発症を抑制するためには自己抗原の認識が重要であることを初めて明らかにしました(Hori et al., PNAS, 2002b; Eur J Immunol, 2002c)。
2001年に帰国し、京都大学再生医科学研究所の坂口志文先生(現大阪大学特別教授)の研究室で、Treg分化と機能の分子基盤という未解明であった本質的な課題に取り組みました。そして、ヒト自己免疫疾患IPEX症候群の原因遺伝子として同定されたFoxp3遺伝子がコードする転写因子Foxp3が、Treg選択的分子マーカーでありその発生・分化と機能を司る“マスター転写因子”として機能することを報告しました(Hori et al., Science, 2003; Hori, Nat Rev Immunol, 2021)。
2004年には、理化学研究所免疫・アレルギー科学総合研究センター(2013年に統合生命医科学研究センターに改組)において独立した研究室を主宰する機会に恵まれました。Foxp3変異マウスに発症する致死的な自己免疫疾患の原因がTregの欠損にあることを明らかにしてTregが「自己」に対する免疫寛容にとって必須の役割を担うことを解明し(Komatsu and Hori, PNAS, 2007)、以下に述べるように、Foxp3を基盤としてTregによる免疫制御メカニズムの解明に取り組んできました。
2016年に東京大学大学院薬学系研究科に異動し、スタッフと学生とともに、それまでの研究テーマを発展させながら、新しい研究テーマを模索しています。
制御性T細胞の運命制御
Tregは、自己抗原や腸内細菌など免疫学的「自己」を構成する物質(抗原)を、T細胞受容体(T cell receptors; TCR)を介して認識することで分化します。
Tregは、免疫応答を抑制する機能に特化した、免疫応答を促進するヘルパーT細胞、キラーT細胞とは独立した細胞系列であり、炎症環境などさまざまな環境に置かれても安定的に免疫抑制機能を維持することで、「自己」に対する免疫寛容を頑健に維持していると考えられています。
そして、この機能的安定性に基づき、Tregを用いた免疫疾患の細胞治療が世界的に試みられています。
我々はFoxp3レポーターマウスを作製して、Foxp3+ T細胞におけるFoxp3発現と抑制機能の安定性を検討しました。
その結果、T細胞欠損マウスへの移入または炎症性サイトカイン存在下でのTCR刺激により、一部のFoxp3+ T細胞がFoxp3発現を失い、抑制活性を持たず炎症や抗体産生を促進するサイトカインを産生するヘルパーT細胞に分化する“可塑性”を示すことを見いだしました(Komatsu et al., PNAS, 2009; Tsuji et al., Science, 2009)。
同様の知見が他のグループからも報告され、Tregが環境変化(特に炎症環境)に応じてヘルパーT細胞に分化転換するという“リプログラミング仮説”が提唱されるようになりました。この現象はTregの機能的安定性とは一見相反しており、Tregが変動する環境にあってどのように免疫寛容を頑健に維持しているのかという根源的な問題を提起しています。
また、Tregは自己反応性を持つため、仮に炎症などの細胞外環境の変化によってヘルパーT細胞にリプログラムされるならば自己免疫疾患の発症を惹起して病態を増悪させることになり、Treg細胞療法の有効性と安全性にも疑問を投げかけることになります。
このため、TregがヘルパーT細胞にリプログラムされ得るのかという問題は世界的な論争を巻き起こしました。
私たちは、Foxp3+ T細胞の可塑性とTregの機能的安定性を矛盾なく説明する枠組みとして、“不均一性モデル”を提唱しました。
すなわち、Foxp3+ T細胞は不均一な細胞集団であり、多くはTregとして運命決定を受けて安定的にFoxp3と抑制機能を発現する一方、Tregとして運命決定を受けていない一部の細胞のみがFoxp3発現を失ってヘルパーT細胞に分化するという考え方です(Hori, Curr Opin Immunol, 2010; Trends Immunol, 2011; Adv Immunol, 2011)。
実際、私たちはFoxp3+ T細胞の運命マッピング解析を通して、生体内に存在するFoxp3+ T細胞は不均一な細胞集団であり、Foxp3発現が誘導されてから間もないCD25-Foxp3+ T細胞が可塑性を示す一方、それ以外の大多数のFoxp3+ T細胞は安定的にFoxp3と抑制機能を発現することを示して“不均一性モデル”を支持する知見を得ました。
また、Foxp3発現を失った細胞のなかには、Foxp3発現を記憶して活性化によって再びFoxp3と抑制機能を発現する“潜在型”Tregが含まれることを見いだしました。そして、TregはFoxp3遺伝子のTSDR(Treg-specific demethylation region)と呼ばれるエンハンサー領域をDNA脱メチル化することによってFoxp3発現を記憶し、安定的にTregに分化した状態を維持していることを明らかにしました(
図1)(Miyao et al., Immunity, 2012)。
私たちはこれら一連の研究を通して、Tregとは細胞外環境の変化に対してもその機能を安定的に維持する細胞運命が決定された細胞系列であり、この“系列安定性”が「自己」に対する免疫寛容の頑健性を保障していること、Foxp3発現のエピジェネティックな記憶こそがTregを規定する本質的なメカニズムであることを提唱しました(Hori, Immunol Rev, 2014)。
これらの知見は、安定的に機能できるTregに分化するためにはFoxp3発現だけでは十分ではなく、TSDR脱メチル化というTreg特有のエピゲノムを獲得することが必要であることを意味しています。Tregエピゲノムの重要性の発見により、2つの本質的な研究課題が生まれ、世界的に研究が進んでいます。第一の課題は、Tregは固有のエピゲノムをどのようなメカニズムで獲得するのか、という問題です。
第二の課題は、環境からのシグナルによりTregエピゲノムはリプログラミングされ得るのかという問題です。私たちは、Foxp3+ T細胞の可塑性はTregのリプログラミングではなく、Foxp3+ T細胞の不均一性により説明できると考えています。しかしながら、現時点では、Tregエピゲノムを単一生細胞レベルで検出し、Foxp3発現を記憶する真のTregの細胞運命を追跡することが技術的に困難であるため、細胞外環境の変化によりTregのエピゲノムが書き換えられてヘルパーT細胞に分化転換し得るのかという問いに対して明確な結論が得られていません。
私たちは、独自の視点とアプローチにより、これらTregの本質を巡るこれら2つの研究課題の解決に取り組んでいます。
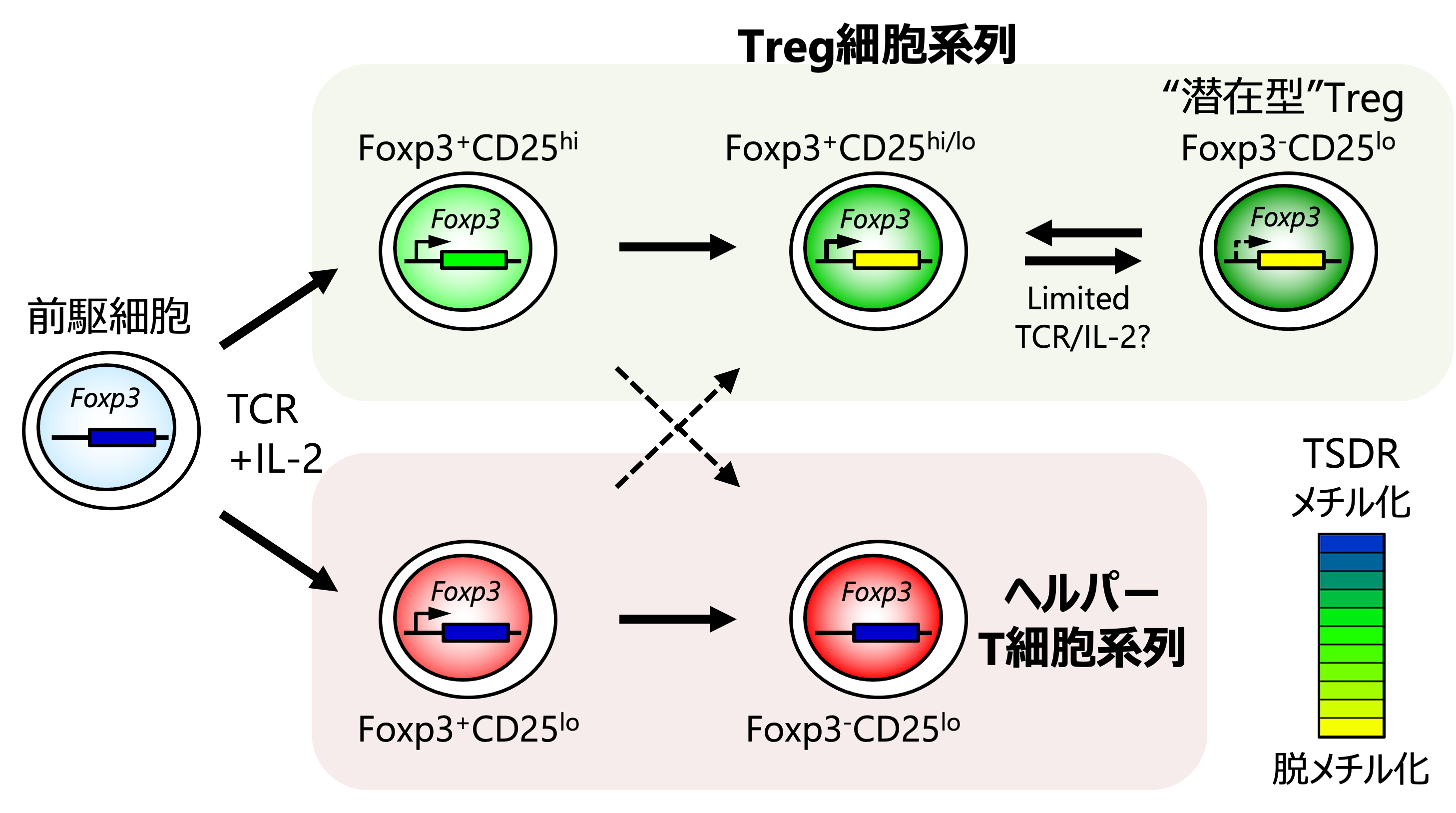
図1
制御性T細胞による適応的免疫制御
Tregは細胞外環境の変化に対して抑制機能を維持しながらも、時々刻々変化する環境に応じて遺伝子発現を柔軟に変化させることにより適応的に免疫応答を制御しています。
Tregのこの環境適応能力(“適応性”adaptability)は、変動する環境においてTregが「自己」に対する免疫寛容と生体の恒常性を維持するために必須の性質であり、その破綻は自己免疫疾患の発症に直結します(Hori and Murakami, Int Immunol, 2021)。
従って、Tregの適応性を保証するメカニズムを解明することは免疫制御システムの動作原理を理解するためのみならず、これを操作してさまざまな疾患を治療するためにも本質的な研究課題です。
私たちはこれまでに、Foxp3によるTreg分化と機能の制御メカニズムを明らかにするために、ヒトIPEX患者で見つかっているFoxp3変異体の機能解析を行ってきました。
特に、DNA結合を担うフォークヘッド・ドメインのミスセンス変異に着目し、変異を導入したマウスモデルを作製して変異がTregと免疫寛容に与える影響を分子レベルから個体レベルにわたって解析してきました。
その過程で、Tregの適応性を特異的に障害することで自己免疫疾患を惹起するFoxp3A384T変異を同定しました。
Foxp3R397W変異やFoxp3I363V変異がDNA結合活性を失わせて標的遺伝子の発現を全体的に障害する機能欠失型変異であるのに対し、Foxp3A384T変異はDNA結合特異性を拡げて一部の標的遺伝子の発現を特異的に障害する機能獲得型変異であることを見いだしました。
そして、Foxp3A384T変異は、Tregが活性化・増殖して抑制機能を発現するエフェクター型Tregに分化し組織に集積する過程を障害して炎症を惹起すること、転写因子BATFの発現抑制がFoxp3A384T変異体によるTregの適応性障害の一因であることを報告しました(図2)(Hayatsu et al., Immunity, 2017)。
興味深いことに、Foxp3A384T変異による影響は、炎症の組織選択性とタイプにおいて特徴的であることがわかりました。Foxp3R397WやFoxp3I363Vなどの機能欠失型変異が皮膚、肺、肝臓などの多臓器にTh1型およびTh2型の炎症を惹起するのに対し、Foxp3A384T変異は皮膚、肺、大腸などの組織においてTregの集積を障害してTh2型およびTh17型の炎症を惹起する一方、肝臓ではTregの集積を障害せず炎症も惹起しません。
このことは、Tregは組織環境、炎症環境からの何らかのシグナルに対して適応的に応答しており、Foxp3A384T変異はその環境由来シグナルに対するTregの適応的な応答を障害するために組織選択的で炎症環境選択的なTregの適応障害を引き起こす可能性を示唆しています。
私たちはこのユニークな発見を手がかりに、Tregによる適応的な免疫制御機構を明らかにすることを目指して研究を進めています。
また、Tregによる適応的免疫制御機構を明らかにするためのもう一つのアプローチとして、Foxp3とTCRシグナルの相互作用に焦点をあてて研究を進めています。
特に、TCRシグナル下流で働く転写因子であるBATFに着目しています。私たちは、Tregが抑制機能を発現するエフェクター型Tregに分化して「自己」に対する免疫寛容を維持するためにBATFが必須であることを明らかにしました(Hayatsu et al., Immunity, 2017 ; Murakami et al., 論文投稿準備中)。
TregはTCRを介して環境に存在する抗原を認識して機能的に活性化するため、BATFはTCRを介した抗原認識をTregの機能発現に結びつける重要な役割を担っていると捉えることができます。一方、Tregの機能を司る“マスター転写因子”がFoxp3です。私たちは、Foxp3がBATFを介してTCRシグナルと相互作用することで、Tregが「自己」に対して機能的に活性化し免疫寛容を維持しているのではないかと考えて研究を進めています。
両者の相互作用メカニズムを明らかにすることができれば、Tregの機能を“抗原特異的に”制御することが可能になると期待できます。そうなれば、免疫寛容を抗原特異的に操作できるようになり、自己免疫疾患や炎症性腸疾患、アレルギー、がんなどのさまざま疾患を特異的に治療できるようになるのではないかと妄想を膨らませています。
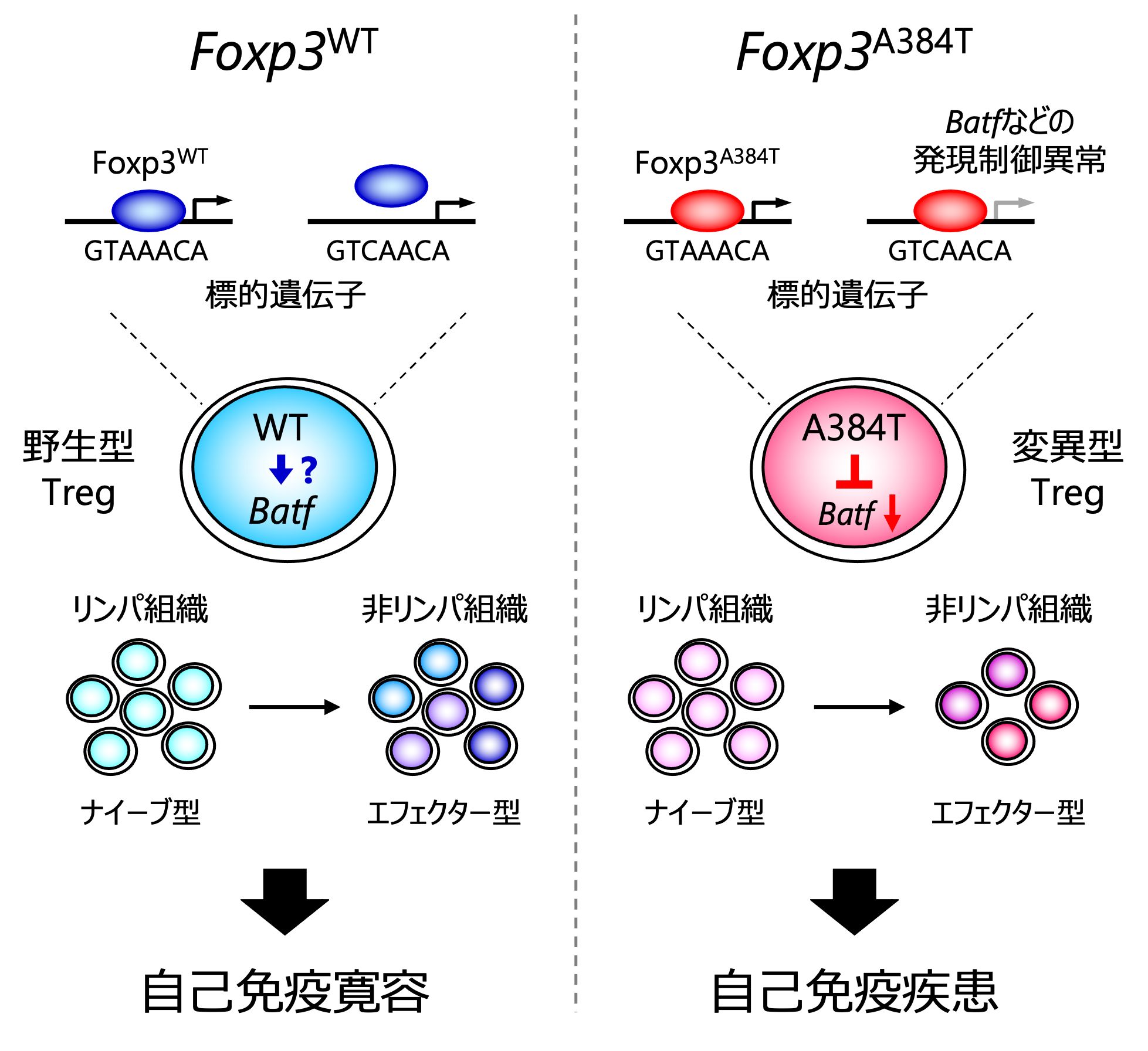
図2
獲得免疫系の動作原理
獲得免疫系最大の特徴は、リンパ球(T細胞とB細胞)集団における2つの異なったレベルの不均一性にあります。一つは抗原受容体(TCRと免疫グロブリン)の遺伝的不均一性です。リンパ球は分化の過程で抗原受容体遺伝子をランダムに再構成することにより膨大な多様性を生み出しています。
そして、この多様性生成の仕組みにより、予測・限定できない多様な物質(抗原)の認識が可能になっています。
もう一つの不均一性は、リンパ球が採る遺伝子発現状態の不均一性(表現型的不均一性)です。リンパ球は細胞外環境からのシグナルにより遺伝子発現を柔軟に変化させることで適切な機能を発現しています。
獲得免疫系がさまざまな疾患から個体を守り、恒常性を維持するためには、個々のリンパ球クローンが、抗原受容体を介して特異的に認識する抗原に適した機能を発現する必要があります。私たちは、獲得免疫においてこれら2つのレベルの不均一性が適切に紐付けられるルール(“紐付けルール”)を解明することが獲得免疫システムの動作原理を解明するうえで本質的な研究課題だと考えています。
そして、さまざまな疾患をこの“紐付けルール”の破綻として理解できるのではないかと考えています(図3)。
私たちは、Tregによる免疫制御機構の研究を通してこの“紐付けルール”の解明に取り組んでいます。Tregの発生・分化と機能には、Foxp3をハブとした遺伝子発現制御ネットワークに加え、TCRを介した「自己」抗原の認識が重要な役割を担っています。
Tregは通常のT細胞と同様に、遺伝的にも表現型的にも不均一な集団であり、多様なTCRレパートリーを持つことで環境に存在するさまざまな抗原に反応し、遺伝子発現を多様に変化させて機能的に異なる状態に分化します。
しかしながら、個々のTregクローンがどのような「自己」抗原を認識することで活性化し、どのように置かれた環境に適した遺伝子発現状態を採り、どのような抗原を認識するT細胞の応答を制御することで「自己」に対する免疫寛容を成り立たせているのか、という問いにはほとんど手が付けられていません。
私たちは、上述のFoxp3A384T変異マウスに発症する組織選択的な自己免疫疾患をモデルとしてこの根源的な問いの解明に取り組んでいます。
すなわち、この変異マウスでは、組織特異的な自己抗原を認識するTregが適切な遺伝子発現状態を採ることができないために(つまりFoxp3A384T変異が遺伝的不均一性と表現型的不均一性の“紐付けルール”を破綻させることにより)組織選択的な自己免疫疾患が発症するのではないかと考えて研究を進めています。
このような独自の視点とアプローチにより、免疫応答を制御する側とされる側のTCRが認識する「自己」の分子的実体を明らかにし、Tregがどのようにして「自己」に対して特異的に免疫寛容を成り立たせているのかを明らかにしたいと考えています。
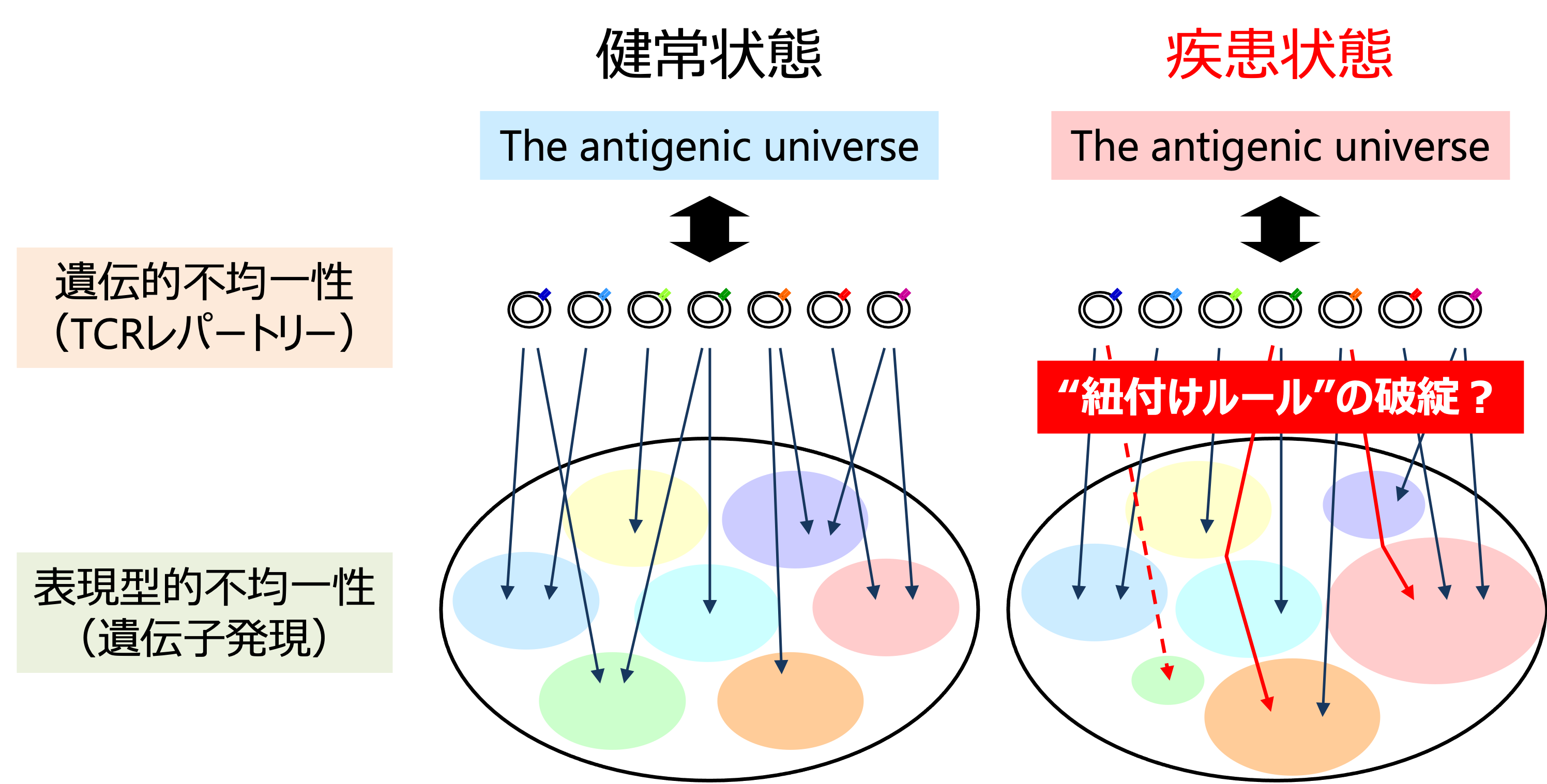
図3
参考文献
- Hori, S., T.L. Carvalho, and J. Demengeot. 2002a. CD25+CD4+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. European Journal of Immunology 32:1282-1291.
- Hori, S., M. Haury, A. Coutinho, and J. Demengeot. 2002b. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 99:8213-8218.
- Hori, S., M. Haury, J.J. Lafaille, J. Demengeot, and A. Coutinho. 2002c. Peripheral expansion of thymus-derived regulatory cells in anti-myelin basic protein T cell receptor transgenic mice. European Journal of Immunology 32:3729-3735.
- Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057-1061.
- Komatsu, N., and S. Hori. 2007. Full restoration of peripheral Foxp3+ regulatory T cell pool by radioresistant host cells in scurfy bone marrow chimeras. Proceedings of the National Academy of Sciences of the United States of America 104:8959-8964.
- Komatsu, N., M.E. Mariotti-Ferrandiz, Y. Wang, B. Malissen, H. Waldmann, and S. Hori. 2009. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proceedings of the National Academy of Sciences of the United States of America 106:1903-1908.
- Tsuji, M., N. Komatsu, S. Kawamoto, K. Suzuki, O. Kanagawa, T. Honjo, S. Hori, and S. Fagarasan. 2009. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science 323:1488-1492.
- Hori, S. 2010. Developmental plasticity of Foxp3+ regulatory T cells. Current Opinion in Immunology 22:575-582.
- Hori, S. 2011a. Regulatory T cell plasticity: beyond the controversies. Trends in Immunology 32:295-300.
- Hori, S. 2011b. Stability of regulatory T-cell lineage. Advances in Immunology 112:1-24.
- Miyao, T., S. Floess, R. Setoguchi, H. Luche, H.J. Fehling, H. Waldmann, J. Huehn, and S. Hori. 2012. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36:262-275.
- Hori S. 2014. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev 259: 159-72.
- Hayatsu, N., T. Miyao, M. Tachibana, R. Murakami, A. Kimura, T. Kato, E. Kawakami, T.A. Endo, R. Setoguchi, H. Watarai, T. Nishikawa, T. Yasuda, H. Yoshida, and S. Hori. 2017. Analyses of a Mutant Foxp3 Allele Reveal BATF as a Critical Transcription Factor in the Differentiation and Accumulation of Tissue Regulatory T Cells. Immunity 47:268-283 e269.
- Hori, S. 2021. FOXP3 as a master regulator of Treg cells. Nat Rev Immunol 21:618-619.
- Hori, S., and R. Murakami. 2021. The adaptability of regulatory T cells and Foxp3. International Immunology 33:803-807.